
Surgical management of hereditary colorectal cancer


Abstract
Colorectal cancer (CRC) is one of the most common solid malignancies worldwide. Although sporadic CRC represents the most common form, genetic alterations is increasingly being identified in a relevant portion of patients with CRC. Familial CRC describes an increased incidence of adenomatous polyps and CRC in first - degree relatives. Hereditary CRC is defined by the identification of deleterious mutations in known predisposing genes. Typical hereditary syndromes with predisposition to CRC include: hereditary non-polyposis colon cancer or Lynch syndrome, familial adenomatous polyposis, attenuated familial adenomatous polyposis, Peutz-Jeghers syndrome and MUTYH associated polyposis. Newly identified genetic alterations with increased risk for CRC include: PPAP, NAD, MSH3 and NTHL1. The diagnosis, surveillance and optimal surgical management of patients with hereditary predisposition to CRC warrant a good understanding of the genetic syndrome in question. Prophylactic surgery must be segregated from symptom-related procedures depending on the syndrome in question. The need for extended surgical procedures must be made in an individualized manner based on gene and gender. The patient should play an active role in the surgical decision-making. Minimally invasive access should be the preferred approach and postoperative quality of life must be seen as a primary outcome measure.
Keywords
Introduction
Colorectal cancer (CRC) represents one of the most common solid malignancies worldwide[1]. Based on the underlying pathogenesis, three distinct groups of CRC can be identified. Sporadic CRC is usually diagnosed in individuals aged 50 years and above and represents approximately 60%-70% of CRC and is as such the most common entity. These patients by definition do not harbor a deleterious predisposing hereditary mutation[2].
The second group has been termed, familial colorectal cancer and describes families with an increased incidence of CRC. Typically, CRC has been diagnosed in more than one close relative. By definition this applies when a first - degree relative (parent, sibling or child) has been diagnosed with CRC[3]. Screening colonoscopy is recommended for the relatives at risk beginning at the age of 40 or 10 years before the youngest age of onset in the family. This group has been reported to account for about 20%-30% of all CRC[3].
The third group includes cases with hereditary CRC, due to a single deleterious mutation in one of the known predisposing genes. Based on multiple mutational abnormalities in different organ systems and additionally a heterogenous phenotype, these mutational pathologies are best summarized as syndromes, which are defined based on clinical, histopathological and genetic findings. With advances in genetic research, it is expected that many more genes involved in predisposition to cancer will be identified in affected individuals.
Well studied hereditary syndromes with predisposition to CRC currently include: hereditary non-polyposis colon cancer (HNPCC) or Lynch syndrome (LS), familial adenomatous polyposis (FAP), attenuated familial adenomatous polyposis (aFAP), Peutz-Jeghers syndrome (PJS) and MUTYH associated polyposis (MAP)[4,5]. Recently described inheritable genetic alterations with increased risk for CRC further include polymerase proofreading associated polyposis (PPAP), NAD, MSH3 and NTH like DNA glycosylase 1 (NTHL1)[5]. All these hereditary syndromes do not only vary widely in terms of clinical presentation but individually present with variable risks of CRC and also involvement of additional benign and malignant neoplastic growth. Interestingly, intrafamilial heterogeneity is common, despite the fact that the affected harbor the identical deleterious mutation. This is an indication of the influence of modifying genetic and epigenetic factors.
The surgical management of patients with hereditary CRC warrants identification and an understanding of the underlying syndrome. The heterogeneity of the phenotype, risk of organ-specific malignant transformation and the quality of life following surgery amongst others must be taken into consideration and discussed at the time of CRC ideally prior to surgery. A personalized approach should always be considered to offer each individual patient the best management option based on gene and gender and the estimated risk, depending on available evidence. However, patient preference is important and the explanation of risk can be challenging.
This review focuses on hereditary syndromes with predisposition to CRC. The most relevant syndromes will be addressed with focus on surgical considerations regarding oncological and functional outcome.
HNPCC (LS)
LS describes a wide spectrum of clinical findings with a high risk of gastrointestinal, urinary and gynecological cancers[6,7]. The underlying pathology is related to defective mismatch repair (MMR) genes, which are associated with a high level of microstatellite instability (MSI-H). These germline mutations are autosomal dominantly transmitted and thus carry a 50% risk of inheritance[8]. The identification of a pathogenic germline MMR mutation (and implicitly a MSI-H tumor phenotype) defines LS[8]. HNPCC is the clinical colorectal manifestation following the familial pattern of inheritance as described in the clinical pattern of the Bethesda or Amsterdam criteria, whereas LS is attributed to an individual with a pathogenic mutation in one of the MMR genes (MLH1, MSH2, MSH6, PMS2, EPCAM).
LS is often associated with a young age of onset and is accountable for 3%-5% of CRC with a predominance in the proximal colon. However, both old age and left-sided or rectal cancers are far more frequent than originally described, since more systematic detection and a more unbiased approach have been pursued. Histopathologically, HNPCC tumors are usually large, mutinous, poorly differentiated and exhibit an extensive lymphocytic infiltration[9]. Despite these unfavorable histopathological features, HNPCC tumors rarely metastasize and are therefore associated with a better prognosis compared to sporadic CRC[10].
The clinical diagnosis of HNPCC is based on the patient’s personal and family history. The Amsterdarm II criteria[11] and the revised Bethesda criteria[12][Table 1] still remain the most common approach for identifying potential Lynch patients or families, despite their poor sensitivity and specificity. Histopathologic examination of tumor biopsies obtained during colonoscopy including immunohistochemical staining for gene products of the MMR genes represents a simple and cost-effective method of identifying individuals with MMR deficiency requiring an individualized therapeutic approach. Of these, approximately 30% will harbour a constitutional mutation, that leads to the diagnosis LS[13]. Therefore all patients with the substantial family history and/or MMR deficiency in the tumor (biopsy) require genetic counseling and if consented genetic testing in a DNA sample in normal tissue.
Amsterdam II criteria and revised Bethesda criteria
| Type | Description |
|---|---|
| Amsterdam II criteria |
|
| Revised Bethesda criteria |
|
Recently, it has been demonstrated that systematic tumor testing without previous staining for loss of protein expression renders a more efficient approach. The authors conclude that “up-front tumor sequencing in colorectal cancer is simpler and has superior sensitivity to current multitest approaches to Lynch syndrome screening, while simultaneously providing critical information for treatment selection”[14]. Also, germline testing in unselected CRC cases with a gene panel [25 known predisposing genes for gastrointestinal tract (GI) cancers] rendered a yield of nearly 10% of identified carriers with a pathogenic mutation. These recent results indicate the need to reassess the value of systematic panel testing either in the tumor or in constitutional DNA as a preferred method for identification of patients with hereditary conditions, rather than staining for MMR deficiency, also taking into account that the polyposis syndromes are almost all stable (exception PPAP and MSH3 polyposis). Specifically for a tailored surgical approach, it is of increasing importance to be aware of the risk disposition for subsequent cancers.
Individuals with a confirmed pathogenic mutation benefit from a regular surveillance program, although the value of each screening procedure must be weighed against potential harm and to date there is a lack of evidence for some of the recommendations. Also, there is no global agreement on the intervals and type of procedures or quality assurance. Beside this, additional data has evolved indicating that a more differentiated approach on the basis of the different MMR genes and gender is required[15].
The indications for colorectal surgery in patients with HNPCC are basically similar to those in patients with sporadic CRC. Premalignant polyps with severe dysplasia, large polyps not amendable via colonoscopy and malignancy represent the most common indications for surgery. LS, in contrast to familial polyposis (FAP) is not associated with full penetrance, therefore prophylactic colorectal resection is not generally recommended. However, the indication for surgery can be extended in selected cases as personalized decision-making based on gene and gender[16-18]. A personalized option might include a combination of (sub)total colectomy in place of the indicated oncological segmental resection at the time of a first CRC in combination with a purely prophylactic hysterectomy with or without adnectomy in a female with LS after completion of family planning.
Surgical management of histologically confirmed LS-associated CRC as a minimal requirement should be in accordance with current oncologic standards. Therefore colon cancer should be managed via colectomy (right or left) without compromise in the oncological thoroughness, despite the observed better prognosis of MMR-deficient tumors. Possibly, complete mesocolic excision (CME) as described by Hohenberger et al.[19] may as is suggested for sporadic cancer demonstrate superiority - this evidence is as of yet not demonstrated. In accordance, cancer of the proximal rectum or rectosigmoid junction should be managed with anterior rectal resection and partial mesorectal excision, whilest mid and low rectal cancers are managed with total mesorectal excision as described by Heald et al.[20]. However, it is mandatory to discuss with patients a prophylactically extended option of additional removal of the colorectum as an alternative to segmental procedures and regular colonoscopies.
The benefits of the laparoscopic approach are well documented for oncological conditions which represent the preferred means of access[21]. For cancer of the mid and lower rectum the transanal total mesorectal excision (taTME) is of increasing importance due to suggested advantages with regard to identification and preservation of pelvic nerves[22,23].
FAP
FAP is the second most common monogenetic hereditary syndrome with predisposition to CRC. FAP is caused by germline mutations of the tumor suppressor APC-gene on chromosome 22q21-22 involving over 2800 codons[24]. The prevalence of this syndrome has been estimated at 1 in 10,000 individuals with the spectrum of clinical presentation depending largely on the mutated codons[25]. Clinically, classical FAP can be distinguished from an attenuated variant (aFAP)[26]. Both conditions are caused by mutations in the same gene, which are allocated to different coding regions.
Classically, FAP is characterized by the development of hundreds to thousands of colonic polyps (adenomas) early in the adolescence[27]. Unlike many other hereditary syndromes with predisposition to CRC, FAP has a one hundred percent penetrance with regard to the development of CRC[28]. Therefore, FAP patients would inevitably develop CRC usually before the age of 40 years if left untreated. More so, close to 13% of FAP patients develop CRC by the age of 25 and about 95% would have developed CRC by the age of 50 years[29].
The attenuated form of FAP (aFAP) is clinically characterized by a later onset on colonic polyposis after ten years compared to cases with classical FAP[30]. Besides, individuals with aFAP present with a significant fewer amount of polyps, usually tens to a few hundreds, mostly in the proximal colon. Unlike FAP, aFAP is not associated with a complete penetrance for CRC. The lifetime risk of CRC in aFAP is estimated at about 70%[31].
FAP (and aFAP) are not only limited to the colon. Extra-colonic manifestations are very common since the germline mutations can affect virtually every organ[32]. Current genetic studies, have identified formerly defined syndromes like Gardner syndrome characterized by FAP, epidermoid cysts, osteomas, desmoid tumors and dental anomalities as merely some aspects of presentation of FAP syndrome[33]. Equally, polyposis of the upper GI is common in FAP patients. Duodenal polyps for example dependent of patient’s age, occur in almost 90% of FAP patients and duodenal cancer is the second most common malignancy in FAP patients[34].
The diagnosis of FAP (aFAP) is usually straight forward. Identifying multiple, up to hundreds of polyps on colonoscopy in an individual with a family history of FAP is de facto the actionable phenotype. Alongside, the presence of extra-colonic manifestations e.g. osteomas, congenital hypertrophy of the retinal pigment epithelium, etc. are indicative of FAP. APC mutation analysis by genetic testing would confirm the suspected diagnosis. However, increasingly rarer heritable syndromes are clinically indistinguishable regarding the colorectal phenotype. In order to adequately judge the risks, it is increasingly important to identify the underlying gene, as demonstrated by gene panel testing with surprising results. This is leading to a shift in paradigm regarding our clinical ability to identify hereditary predispositions to cancer.
Individuals with genetically confirmed FAP as well as individuals at risk should undergo endoscopic surveillance every 1-2 years. The recommended surveillance frequency increases to once per year as soon as adenomatous polyps appear. This interval stays until surgical management via proctocolectomy (colectomy for aFAP) is performed and is further warranted for manifestations in other organs[35].
Generally accepted indications for surgical management include large adenomas > 1 cm, numerous adenomas (> 20), unfavorable histology (high grade dysplasia or carcinoma), chronic anemia and failure to thrive. Otherwise, surgical management is performed in a personalized manner, usually if possible postponed until after puberty[36].
Restorative proctocolectomy with an ileal pouch anal anastomosis (IPAA) is the recommended treatment for classical FAP. The vulnerable colon and rectum are removed leaving a 2 cm sensitive rectal cuff (transition zone) above the anus for solid-liquid-gas discrimination[37]. The rectal mucosal cuff, however, must be seen as a risky island with a residual risk of mucosal dysplasia as high as 4.5% after 10 years follow-up[38]. Thus the need for a stringent endoscopic follow-up cannot be over emphasized. Some surgeons perform a mucosectomy during IPAA with the goal of preventing future rectal cuff cancer although poor functionality has been reported following mucosectomy[39]. This has been attributed primarily to loss of the sensitive transitional zone and secondary to injury of the sphincter apparatus during dissection. We perform mucosectomy in selected cases when the transitional zone is involved in the disease process at the time of primary surgery. Also, the recurrence rate of neoplasia following primary mucosectomy remains high and may be repeated. For this reason at our institution we have also implemented the taTME approach for benign disease. By performing a double purse-string anastomosis at the height of the dentate line the rectal remnant and all mucosa is eliminated, it may be expected that this will further reduce the neoplastic changes seen today in the ageing FAP population with a rectal remnant of (at least) 2 cm. Functional results in this small series, to date are excellent but require prospective documentation.
Preserving the rectum and it’s reservoir function via a colectomy with an ileorectal anastomosis is also an established option in most aFAP individuals. This option is open to individuals with little or no rectal polyposis. The advantages of the ileorectal anastomosis include a reduced number of bowel movements, a better continence and potentially a better quality of life compared to IPAA, although this has not been proven[40]. However, such patients need to be closely monitored (annually initially, then in 4-6 monthly intervals after the age of 50) via endoscopy of the disposed rectal remnant. Subtotal colectomy with ileosigmoid anastomosis should be considered in individuals with aFAP with polyposis of the proximal colon. These options should be considered in the attenuated FAP phenotype and especially in female patients of reproductive age to avoid pelvic dissection and formation of adhesions.
Surgical management of individuals with FAP (and aFAP) should preferably be done via minimally invasive access. Laparoscopic colectomy and proctocolectomy now represent standard procedures for FAP patients. An increasing number of FAP patients are developing desmoid tumors[41]. These fibrous tumors are considered to be triggered by trauma, including surgical trauma. Thus reducing surgical trauma via laparoscopic access should be a primary goal in these patients. This is also true with regard to the creation of a diverting ileostomy during IPAA. Ileostomy creation and eventually reversal contribute to significant surgical trauma with an increased risk of desmoid tumor. We routinely use a virtual ileostomy (ghost ileostomy) [Figure 1] during IPAA for FAP instead of a diverting ileostomy[42].
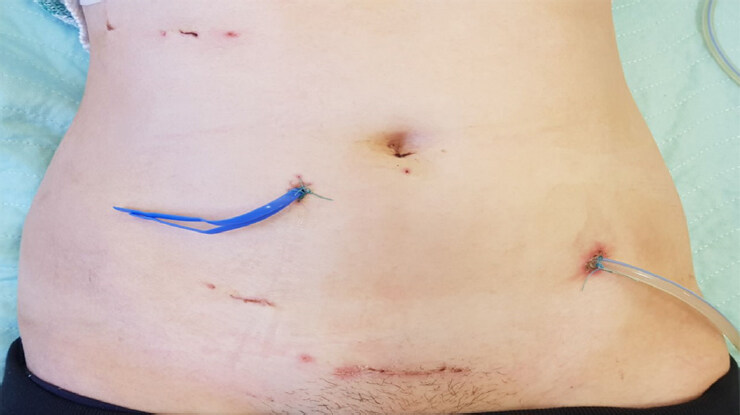
Figure 1. Virtual ileostomy with an exteriorized blue vassel loop following ileal pouch anal anastomosis for familial adenomatous polyposis
A majority of patients undergoing prophylactic proctocolectomy with FAP have no evidence of cancer. Therefore, oncologic dissection must not be performed in these patients. However, we prefer central dissection of the mesentery for three reasons: first central dissection with CME is standard for oncologic segmental colectomy. We therefore have expertise in this dissection, which is time-sparing with less vessel ligations required. Second, there is a possibility that cancer might be found in the surgical specimen for which oncologic resection would have been indicated. Thus a preemptive oncologic resection is sensible. Third, the mesenteric remnant is a predisposing site for the development of desmoid tumors [Figure 2]. In this light, pelvic dissection should be performed in accordance with TME because desmoid tumors in the remaining rectal mesentery may later impair pouch functionality. These surgical aspects are of utmost importance because desmoid tumors comprise the second most common cause of death after cancer in patients with FAP and are the most relevant factor deteriorating the quality of life in FAP patients.
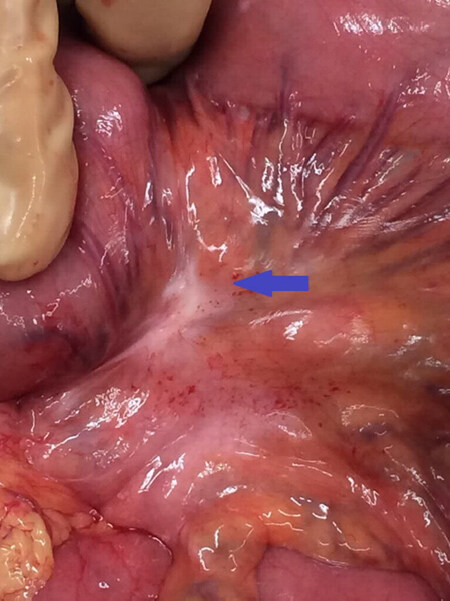
Figure 2. The arrow indicates a desmoid plaque of the small bowel mesenterium
Patients undergong (prophylactic) restorative proctocolectomy with IPAA for FAP are usually young and active. Therefore, quality of life is a central outcome measure in these patients. Key parameters in this regard are the number of bowel movements and fecal continence. The continent function depends mainly on the stool consistency, but also on the intactness of both the sphincter apparatus and the pelvic nerves. The identification and preservation of these nerves could be difficult via the top - bottom dissection during laparoscopic surgery. TaTME is now an established method in the management of mid and low rectal cancer[43]. This technique is a further modification of transanal minimally invasive techniques and enables a bottom - top dissection with improved visualization of the pelvic nerves and a rendezvous-approach. TaTME now represents our standard procedure for pelvic dissection during proctocolectomy[44].
The anastomosis technique for IPAA remains an issue of controversial debate. The most common anastomosis techniques include the double purse - string with single stapling, double stapling and the hand-sewn anastomosis. The double stapling technique is easy to perform. However, stapler intersection might predispose to anastomotic dehiscence, which is the most severe complication following IPAA. The hand-sewn anastomosis can be challenging and demanding in unexperienced hands. Continence function has been shown to be better following double stapling in comparison to hand-sewn anastomosis[45].
The single - stapling technique following double purse-string is our preferred technique for IPAA. Outcomes with this technique are similar to the double-stapled technique. A major advantage of the single stapled anastomosis is the lack of interposition of staplers predisposing to anastomotic dehiscence.
The need of a diverting ileostomy in patients undergoing IPAA for FAP remains a point of controversy. FAP patients are usually young and otherwise healthy with no risk factors for anastomotic leakage[46]. The rate of anastomotic leakage as high as 10% has been reported in this population[47]. Due to the fear of devastating consequences following anastomotic leakage, some surgeons prefer to perform fecal diversion during IPAA. We generally do not routinely use a diverting ileostomy during IPAA. Instead we use a virtual ileostomy [Figure 1], which can be easily converted to a defunctional ileostomy in case of a clinically relevant anastomotic leakage[42]. We complete the procedure with the placement of a transanal decompression tube [Figure 3].
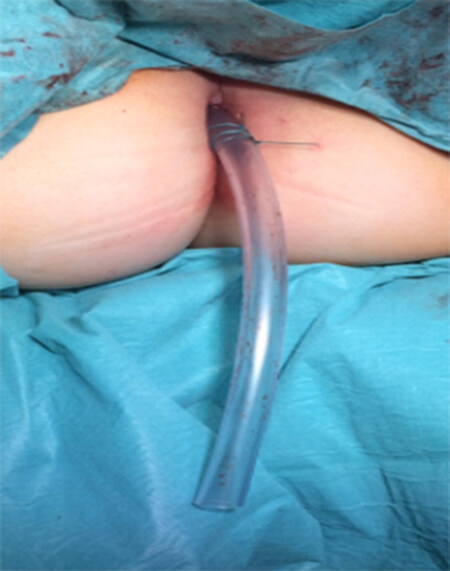
Figure 3. Placement of a transanal decompression tube into the ileal pouch at the end of ileal pouch anal anastomosis
MAP
MAP is caused by biallelic mutations in the MUTYH (myh) gene. The MUTYH gene is involved in base excision repair in the set-up of oxidative DNA damage by preventing G:C to T:A transversions[48]. Unlike monoallelic mutations with predisposition to CRC with an autosomal dominant transmission, siblings of biallelic MUTYH mutations have a 25% chance of having MAP. This is based on the autosomal recessive transmission pattern, indicating that parents and children of affected individuals are usually not affected[49].
The clinical presentation in MAP is similar to that of aFAP, with tens to hundreds of polyps with proximal colonic predominance by the age of 40-45 years found during colonoscopy[50]. Although adenomatous polyps predominate, hyperplasic polyps are common in MAP[51]. This is a major distinguishing histologic feature. Genetic testing for MAP is recommended in patients with clinical features of FAP or aFAP without identifiable APC mutation. The gene is included in all gene panels for hereditary GI predisposition and is not infrequent in this setting.
Colonoscopic surveillance is recommended in individuals with MAP typically starting in their mid-20’s. There is no indication for prophylactic surgery without neoplastic changes. Subtotal colectomy is the procedure of choice in MAP patients with CRC or severe dysplasia or a high polyp burden. Relative indications for surgery include troublesome colonoscopy or the situations when polyps become too large to be removed endoscopically.
Hamartomatous polyposis syndromes
Peutz-jeghers syndrome (PJS) is a genetic syndrome associated with hamartomatous polyposis with an increased risk of CRC and other malignancies. Similar syndromes with predisposition to colonic polyposis include juvenile polyposis syndrome (JPS) and Cowden syndrome. PJS is caused by mutations in STK11 - gene[52]. Mutations in SMAD4 and BMPR1A genes have been identified in 40%-60% of patients with JPS[53,54]. Mutations in the PTEN gene are found in over 80% of patients with Cowden syndrome[55].
One of the most characteristic feature of PJS is the development of multiple hamartomatous polyps of both the small (60%-90%) and large (50%-70%) intestines[56]. Symptoms usually occur in teenage years, however in our series children with polyp-related intussusception and emergency surgery under the age of eight years much higher than anticipated (5% of all PJS patients). Intussusception, gastrointestinal bleeding and bowel obstruction are commonly the first clinical symptoms, apart from the lentiginosis pigmentosa of the lips[57].
Mucocutaneous pigmentation on the lips, periorbital region and buccal mucosa are very consistent in patients with PJS. This mucocutaneous pigmentation in association with hamartomatous polyps are virtually diagnostic for PJS in an individual with a family history of PJS[58,59]. The lifetime risk of cancer development in patients with PJS has been reported to be as high as 85%[60]. Cancers of the GI have been thought to occur in about three quarters of PJS patients including a close to 40% risk for CRC[61].
Since PJS is not associated with complete penetrance with regard to CRC there is no indication for prophylactic surgery. The indications for surgical management are therefore usually symptom related. This is especially true for surgical management of small bowel polyps. The small bowel must be monitored regularly, preferably with video endoscopy and polyps with a size > 1.5 cm should be prophylactically removed, preferably via doubl-balloon endoscopy. Surgical resection of large colonic polyps is indicated following failure of endoscopic polypectomy. Equally, severe dysplasia and malignant transformation constitute absolute indications for surgery. The principles of oncologic resection should be followed when dealing with CRC in a patient with PJS.
PPAP
PPAP is a dominantly inherited condition caused by germline mutations in the DNA polymerases POLE and POLD[62]. These mutations have been identified in families with a history of unexplained adenomatous polyposis and CRC[63,64]. Extra-colonic manifestations including endometrial, brain and duodenal tumors have been reported in association with PPAP[65]. Typically, a large number of polyps (up to a few hundreds) are present by the age of 35-40 years[66]. Therefore, PPAP should be considered if clinical and endoscopic aspects of FAP, aFAP or MAP are present in the absence of the respective mutations following genetic analysis. Also and interestingly, this syndrome may clinically be consistent with LS, including MSI in the neoplastic tissue.
Estimated risks of PPAP - associated CRC patients with POLE mutations by the age of 70 years were reported in a recent publication by Buchanan et al.[67] to be 40% for males and 32% for females. The corresponding risks in patients with POLD mutations were 63% in males and 52% in females. These estimates must be interpreted with caution due to limited data on this newly described syndrome.
An evidence based management algorithm for PPAP is so far not available. Thus surveillance and surgical management of patients with PPAP should follow the principles of “best clinical practice” analogue to similar syndroms (aFAP and MAP and LS).
NTHL1-associated polyposis
NTHL1-associated polyposis (NAP) is a recessively inherited polyposis syndrome caused by mutations in base excision repair gene NTHL1[68]. Homozygous germline mutations in NTHL1 have been identified in cases with a family history of adenomatous polyposis and CRC[69]. Besides, extra-colonic manifestations might be present[70]. More insight is needed to be able to define the spectrum of presentation and the risk of CRC in patients with NAP. To date, surgical management is symptom related, with no indication for prophylactic surgery at this time.
MSH3 polypsis
Adenomatous polyposis has recently been described in connection with biallelic germline mutations in MSH3, an MMR gene[71]. The resulting CRC demonstrates microsatellite instability. Although data on this new polyposis syndrome are limited, surveillance and management should follow the principles of “best clinical practice” analogue to LS. Clinically, the phenotype might be indistinguishable from FAP and follow the same (individualized) management recommendations.
Discussion
The management of patients with hereditary predisposition for CRC warrants a good understanding of the underlying syndrome. The syndrome-associated risk for CRC must be considered, especially in the event of a CRC and required surgery. Besides, heterogeneity in mutational status as well as clinical presentation must be considered in each case. Thus an individualized decision-making and a personalized strategy with the patient’s active involvement in terms of shared decision-making should represent a major aspect of management.
Basically, the principles of oncologic surgery should be respected in call cases with CRC independent of mutational status. The need for extended surgery beyond segmental colectomy for CRC can be made as a “tailored approach” in selected cases based on mutational status, that is gene and gender. This is a moving target and the challenge lies in the translational aspect of patient management. As an example: LS still may be considered by most as a syndrome with a high risk for colorectal and other GI malignancies. However, depending on the gene, endometrial cancer may be the sentinel cancer and not CRC (MSH6). Or, PMS2 is a very low penetrant gene and mutations may not predispose to a substantial amount of cancers. Therefore annual invasive screening or prophylactic surgery may not be warranted. MLH1- and MSH2-LS patients, however, have a very high risk for metachronous CRC cancers, despite even yearly colonoscopies. Therefore, patients might opt to have more extended surgery at the time of their primary surgery. Or, especially for MSH6 mutation carriers, a simultaneous hysterectomy after completion of the family planning might be the preferred option. In order to address these issues it is becoming pivotal to generate genetic tests and a reliable risk assessment as timely as possible following the diagnosis of CRC and prior to cancer surgery.
Prophylactic colorectal resection (restorative proctocolectomy) with or without IPAA is currrently reserved for hereditary syndroms with 100% penetrance for the development of CRC including FAP, aFAP and MAP. The role of prophylactic colorectal surgery for hereditary syndromes without a 100% penetrance is still to be defined and must include patient preference.
Patients with hereditary CRC are usually younger than those with sporadic CRC. Thus quality of life following surgery is of even greater importance to these patients. Therefore attention to technical details with respect to surgery like minimizing surgical trauma and preservation of nerve function during pelvic dissection is a major aspect of surgical management. Furthermore, the postoperative follow-up should be in accordance with syndrome-specific guidelines.
The role of effective chemoprevention for example with aspirin may influence decision-making regarding prophylactic surgery and must be assessed prospectively. In the light of evolving evidence, it is mandatory to involve patients in decision-making with the most recent knowledge available.
In conclusion, with increasing understanding of tumor genetics, the role of gene and gender will increasingly play a role in the management of patients with hereditary predispostion for CRC. A good understanding of the predisposing genetic mutations with regard to mutational and clinical heterogeneity is the basis for an optimized management. Prophylactic surgery is reserved for syndromes with a 100% penetrance. The principles of oncologic resection should be respected as in sporadic CRC. The quality of life following surgical management should be the focus of counselling and decision-making. Postoperative lifelong follow-up is an important aspect of surgical management and must be discussed with patients at the time of their first surgery. The potential role of chemoprevention in individuals with hereditary predisposition to CRC must be prospectively studied and evaluated for less invasive options than prophylactic or prophylactically extended surgeries. Last but not least: after identification of an index patient with a hereditary mutation, genetic counselling and the option of predictive testing must be the focus of the surgeon.
Declarations
Authors’ contributionsDesign, literature research and review, critically reviewed the manuscript, final approval: Ambe PC, Möslein G
Drafted the manuscript: Ambe PC
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. International Agency for Research on Cancer. World cancer report 2014. Available from: https://www.drugsandalcohol.ie/28525/1/World%20Cancer%20Report.pdf. [Last accessed on 17 Oct 2018].
2. Gamboa A, Lin Q, Jin P, Zhou Y, Liu Q, et al. The descriptive epidemiology of gastrointestinal malignancies. In: Cai Q, El-Rayes BF, Hao J, editors. Gastrointestinal malignancies: new innovative diagnostics and treatment. New Jersey: World Scientific; 2015. pp. 1-40.
3. Lung MS, Trainer AH, Campbell I, Lipton L. Familial colorectal cancer. Intern Med J 2015;45:482-91.
5. Lorans M, Dow E, Macrae FA, Winship IM, Buchanan DD. Update on hereditary colorectal cancer: improving the clinical utility of multigene panel testing. Clin Colorectal Cancer 2018;17:e293-305.
6. Lynch HT, Lynch J. Lynch syndrome: genetics, natural history, genetic counseling, and prevention. J Clin Oncol 2000;18:19S-31S.
7. Planck M1, Rambech E, Möslein G, Müller W, Olsson H, et al. High frequency of microsatellite instability and loss of mismatch-repair protein expression in patients with double primary tumors of the endometrium and colorectum. Cancer 2002;94:2502-10.
8. Papadopoulos N, Lindblom A. Molecular basis of HNPCC: mutations of MMR genes. Hum Mutat 1997;10:89-99.
9. Shia J, Ellis NA, Paty PB, Nash GM, Qin J, et al. Value of histopathology in predicting microsatellite instability in hereditary nonpolyposis colorectal cancer and sporadic colorectal cancer. Am J Surg Pathol 2003;27:1407-17.
10. Malesci A, Laghi L, Bianchi P, Delconte G, Randolph A, et al. Reduced likelihood of metastases in patients with microsatellite-unstable colorectal cancer. Clin Cancer Res 2007;13:3831-9.
11. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the international collaborative group on HNPCC. Gastroenterology 1999;116:1453-6.
12. Rodriguez-Bigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR, et al. A national cancer institute workshop on hereditary nonpolyposis colorectal cancer syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst 1997;89:1758-62.
13. Müller W, Burgart LJ, Krause-Paulus R, Thibodeau SN, Almeida M, et al. The reliability of immunohistochemistry as a prescreening method for the diagnosis of hereditary nonpolyposis colorectal cancer (HNPCC)--results of an international collaborative study. Fam Cancer 2001;1:87-92.
14. Hampel H, Pearlman R, Beightol M, Zhao W, Jones D, et al. Assessment of tumor sequencing as a replacement for Lynch syndrome screening and current molecular tests for patients with colorectal cancer. JAMA Oncol 2018;4:806-13.
15. Vasen HF, Blanco I, Aktan-Collan K, Gopie JP, Alonso A, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut 2013;62:812-23.
16. Rau B, Angele M, Gonzalez-Moreno S, Kähler G, Langelotz C, et al. Gender-specific aspects in gastrointestinal medicine and surgery. Viszeralmedizin 2014;30:133-5.
17. Möslein G, Ohmann C, Wenzel M. Prophylactic surgery for hereditary non-polyposis colorectal cancer. Chirurg 2005;76:1135-44. (in German)
18. Schneider R, Fürst A, Möslein G. Gender-specific aspects of Lynch syndrome--an update. Z Gastroenterol 2015;53:789-93. (in German)
19. Hohenberger W, Weber K, Matzel K, Papadopoulos T, Merkel S. Standardized surgery for colonic cancer: complete mesocolic excision and central ligation--technical notes and outcome. Colorectal Dis 2009;11:354-64.
20. Heald RJ, Moran BJ, Ryall RD, Sexton R, MacFarlane JK. Rectal cancer: the Basingstoke experience of total mesorectal excision, 1978-1997. Arch Surg 1998;133:894-9.
21. Hazebroek EJ; Color Study Group. COLOR: a randomized clinical trial comparing laparoscopic and open resection for colon cancer. Surg Endosc 2002;16:949-53.
22. Kneist W, Hanke L, Kauff DW, Lang H. Surgeons’ assessment of internal anal sphincter nerve supply during TaTME - inbetween expectations and reality. Minim Invasive Ther Allied Technol 2016;25:241-6.
23. Kneist W, Wachter N, Paschold M, Kauff DW, Rink AD, et al. Midterm functional results of taTME with neuromapping for low rectal cancer. Tech Coloproctol 2016;20:41-9.
24. Aretz S, Uhlhaas S, Sun Y, Pagenstecher C, Mangold E, et al. Familial adenomatous polyposis: aberrant splicing due to missense or silent mutations in the APC gene. Hum Mutat 2004;24:370-80.
25. Bisgaard ML, Fenger K, Bülow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum mutat 1994;3:121-5.
26. Knudsen AL, Bülow S, Tomlinson I, Möslein G, Heinimann K, et al. Attenuated familial adenomatous polyposis: results from an international collaborative study. Colorectal Dis 2010;12:e243-9.
28. Dörner J, Fallahpour MT, Möslein G. Surgical management of hereditary colorectal cancer syndromes. In: Valle L, Gruber S, Capellá G, editors. Hereditary colorectal cancer. Cham: Springer; 2018. pp. 327-47.
29. De Cosse JJ, Bülow S, Neale K, Järvinen H, Alm T, et al. Rectal cancer risk in patients treated for familial adenomatous polyposis. The Leeds Castle polyposis group. Br J Surg 1992;79:1372-5.
30. Lynch HT, Smyrk T, McGinn T, Lanspa S, Cavalieri J, et al. Attenuated familial adenomatous polyposis (AFAP). A phenotypically and genotypically distinctive variant of FAP. Cancer 1995;76:2427-33.
31. Knudsen AL, Bisgaard ML, Bülow S. Attenuated familial adenomatous polyposis (AFAP): a review of the literature. Fam cancer 2003;2:43-55.
32. Groen EJ, Roos A, Muntinghe FL, Enting RH, de Vries J, et al. Extra-intestinal manifestations of familial adenomatous polyposis. Ann Surg Oncol 2008;15:2439-50.
33. Traboulsi EI, Krush AJ, Gardner EJ, Booker SV, Offerhaus GJ, et al. Prevalence and importance of pigmented ocular fundus lesions in Gardner’s syndrome. N Engl J Med 1987;316:661-7.
34. Naylor EW, Gardner EJ, Richards RC. Desmoid tumors and mesenteric fibromatosis in Gardner’s syndrome: report of kindred 109. Arch Surg 1979;114:1181-5.
35. Vasen HF, Möslein G, Alonso A, Aretz S, Bernstein I, et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut 2008;57:704-13.
36. Lynch PM, Morris JS, Wen S, Advani SM, Ross W, et al. A proposed staging system and stage-specific interventions for familial adenomatous polyposis. Gastrointest Endosc 2016;84:115-25.
38. Remzi FH, Fazio VW, Delaney CP, Preen M, Ormsby A, et al. Dysplasia of the anal transitional zone after ileal pouch-anal anastomosis: results of prospective evaluation after a minimum of ten years. Dis Colon Rectum 2003;46:6-13.
39. Carne PW, Pemberton JH. Technical aspects of ileoanal pouch surgery. Clin Colon Rectal Surg 2004;17:35-41.
40. Van Duijvendijk P, Slors JF, Taat CW, Oosterveld P, Sprangers MA, et al. Quality of life after total colectomy with ileorectal anastomosis or proctocolectomy and ileal pouch-anal anastomosis for familial adenomatous polyposis. Br J Surg 2000;87:590-6.
41. DE Marchis ML, Tonelli F, Quaresmini D, Lovero D, Della-Morte D, et al. Desmoid tumors in familial adenomatous polyposis. Anticancer Res 2017;37:3357-66.
42. Ambe PC, Zirngibl H, Möslein G. Routine virtual ileostomy following restorative proctocolectomy for familial adenomatous polyposis. World J Surg 2018;42:1867-71.
43. Monson JR, Arsalanizadeh R. Transanal total mesorectal excision (TaTME) and quality of rectal cancer surgery: do we really know? Ann Surg 2017;266:e88-9.
44. Ambe PC, Zirngibl H, Möslein G. Initial experience with taTME in patients undergoing laparoscopic restorative proctocolectomy for familial adenomatous polyposis. Tech Coloproctol 2017;21:971-4.
45. Reilly WT, Pemberton JH, Wolff BG, Nivatvongs S, Devine RM, et al. Randomized prospective trial comparing ileal pouch-anal anastomosis performed by excising the anal mucosa to ileal pouch-anal anastomosis performed by preserving the anal mucosa. Ann Surg 1997;225:666-76.
46. Rullier E, Laurent C, Garrelon JL, Michel P, Saric J, et al. Risk factors for anastomotic leakage after resection of rectal cancer. Br J Surg 1998;85:355-8.
47. Aziz O, Albeyatti A, Derias M, Varsani N, Ashrafian H, et al. PTU-202 A case-controlled study demonstrating that changes associated with rectal anastomotic leakage are detectable within 48 hours of surgery. Gut 2015;64:A152.
48. Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, et al. Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat Genet 2002;30:227-32.
50. Nielsen M, Joerink-van de Beld MC, Jones N, Vogt S, Tops CM, et al. Analysis of MUTYH genotypes and colorectal phenotypes in patients With MUTYH-associated polyposis. Gastroenterology 2009;136:471-6.
51. Nieuwenhuis MH, Vogt S, Jones N, Nielsen M, Hes FJ, et al. Evidence for accelerated colorectal adenoma--carcinoma progression in MUTYH-associated polyposis? Gut 2012;61:734-8.
52. De Rosa M, Galatola M, Quaglietta L, Miele E, De Palma G, et al. Alu-mediated genomic deletion of the serine/threonine protein kinase 11 (STK11) gene in Peutz-Jeghers syndrome. Gastroenterology 2010;138:2558-60.
53. Howe JR, Roth S, Ringold JC, Summers RW, Järvinen HJ, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 1998;280:1086-8.
54. Howe JR, Bair JL, Sayed MG, Anderson ME, Mitros FA, et al. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet 2001;28:184-7.
55. Orloff MS, Eng C. Genetic and phenotypic heterogeneity in the PTEN hamartoma tumour syndrome. Oncogene 2008;27:5387-97.
56. Utsunomiya J, Gocho H, Miyanaga T, Hamaguchi E, Kashimure A. Peutz-Jeghers syndrome: its natural course and management. Johns Hopkins Med J 1975;136:71-82.
57. Beggs AD, Latchford AR, Vasen HF, Moslein G, Alonso A, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut 2010;59:975-86.
58. Jeghers H, McKusick VA, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med 1949;241:993.
59. Giardiello FM, Trimbath JD. Peutz-Jeghers syndrome and management recommendations. Clin Gastroenterol Hepatol 2006;4:408-15.
60. Hearle N, Schumacher V, Menko FH, Olschwang S, Boardman LA, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res 2006;12:3209-15.
61. Lefevre H, Bouvattier C, Lahlou N, Adamsbaum C, Bougnères P, et al. Prepubertal gynecomastia in Peutz-Jeghers syndrome: incomplete penetrance in a familial case and management with an aromatase inhibitor. Eur J Endocrinol 2006;154:221-7.
62. Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 2013;45:136-44.
63. Spier I, Holzapfel S, Altmüller J, Zhao B, Horpaopan S, et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int J Cancer 2015;137:320-31.
64. Valle L, Hernández-Illán E, Bellido F, Aiza G, Castillejo A, et al. New insights into POLE and POLD1 germline mutations in familial colorectal cancer and polyposis. Hum Mol Genet 2014;23:3506-12.
65. Hahn MM, Vreede L, Bemelmans SA, van der Looij E, van Kessel AG, et al. Prevalence of germline mutations in the spindle assembly checkpoint gene BUB1B in individuals with early-onset colorectal cancer. Genes Chromosomes Cancer 2016;55:855-63.
66. Bellido F, Pineda M, Aiza G, Valdés-Mas R, Navarro M, et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet Med 2016;18:325-32.
67. Buchanan DD, Stewart JR, Clendenning M, Rosty C, Mahmood K, et al. Risk of colorectal cancer for carriers of a germ-line mutation in POLE or POLD1. Genet Med 2018;20:890-5.
68. Weren RD, Ligtenberg MJ, Kets CM, de Voer RM, Verwiel ET, et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet 2015;47:668-71.
69. Broderick P, Dobbins SE, Chubb D, Kinnersley B, Dunlop MG, et al. Validation of recently proposed colorectal cancer susceptibility gene variants in an analysis of families and patients-a systematic review. Gastroenterology 2017;152:75-7.
70. Chubb D, Broderick P, Dobbins SE, Frampton M, Kinnersley B, et al. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat Commun 2016;7:11883.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Ambe PC, Möslein G. Surgical management of hereditary colorectal cancer. Mini-invasive Surg 2018;2:37. http://dx.doi.org/10.20517/2574-1225.2018.45
AMA Style
Ambe PC, Möslein G. Surgical management of hereditary colorectal cancer. Mini-invasive Surgery. 2018; 2: 37. http://dx.doi.org/10.20517/2574-1225.2018.45
Chicago/Turabian Style
Ambe, Peter C., Gabriela Möslein. 2018. "Surgical management of hereditary colorectal cancer" Mini-invasive Surgery. 2: 37. http://dx.doi.org/10.20517/2574-1225.2018.45
ACS Style
Ambe, PC.; Möslein G. Surgical management of hereditary colorectal cancer. Mini-invasive. Surg. 2018, 2, 37. http://dx.doi.org/10.20517/2574-1225.2018.45
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 24
likes
Like This Article 24
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.